Síndrome coronario agudo sin elevación del ST (SCASEST) de alto riesgo
como manifestación clínica grave del síndrome de Cogan
[High-risk non-ST-segment elevation acute coronary syndrome as an acute
clinical manifestation of Cogan syndrome]
Lucas Barros López,* Jessica Giménez Lecina, Mónica Requesens Solera, Sara Noblejas Drouot,
María Barrera Sánchez, José Javier Muñoz Marcos
Servicio de Medicina Intensiva, Hospital Universitario Miguel Servet,
Zaragoza, España
* Correspondencia: lucas_bl6@hotmail.com
Los
autores no declaran conflictos de intereses.
Resumen
El
síndrome de Cogan es un
cuadro inflamatorio poco frecuente que habitualmente se presenta con
síntomas oculares y vestibulococleares, pero que, en un bajo porcentaje de los
casos, puede asociarse con manifestaciones sistémicas graves que se asemejan a
una vasculitis. Su tratamiento no está claro y se basa en regímenes
inmunosupresores similares a los administrados para otras enfermedades
autoinmunes. Presentamos el caso de una paciente con síndrome de Cogan y
aortitis que causaba la compresión extrínseca del tronco coronario izquierdo,
su evolución y tratamiento. Asimismo, el objetivo de este artículo es analizar
la evidencia científica disponible sobre casos similares con manifestaciones
sistémicas que ponen en riesgo la vida.
Palabras clave: Síndrome de Cogan; vasculitis; aortitis;
síndrome vestibular.
Abstract
Cogan syndrome is a rare inflammatory condition that typically presents
with ocular and vestibulocochlear symptoms, but in a small percentage of cases,
it can be associated with serious systemic manifestations resembling
vasculitis. The treatment of this syndrome is not clear and is based on
immunosuppressive regimens similar to those of other autoimmune diseases. We
present the case of a patient with Cogan syndrome and aortitis, which caused
extrinsic compression of the left coronary trunk, its progression and
treatment. The aim of this article is to review the available scientific evidence
on similar cases with systemic manifestations that carry life-threatening
risks.
Keywords: Cogan syndrome; vasculitis; aortitis;
vestibular syndrome.
Introducción
El síndrome
de Cogan, una enfermedad inflamatoria poco común, fue identificado por primera
vez, en 1945, por el oftalmólogo David Cogan como una causa de queratitis
intersticial no sifilítica acompañada de síntomas vestibulococleares.
Esta revisión
tiene como objetivo proporcionar un entendimiento detallado de las
características clínicas del síndrome, sus desafíos diagnósticos y las opciones
de tratamiento disponibles.
Caso clínico
Mujer de 38
años con síndrome de Cogan de tres años de evolución. El síndrome había sido
diagnosticado a raíz del compromiso neurovestibular severo que requirió un
implante coclear derecho, además, de síndrome vertiginoso crónico, queratitis
intersticial y policondritis recidivante. La paciente tenía manifestaciones
clínicas desde el diagnóstico sugerentes de vasculitis de pequeño y grandes
vasos, con compromiso de la aorta ascendente, el cayado aórtico, la subclavia
izquierda, la aorta abdominal y estenosis de ilíacas primitivas.
En el momento
del diagnóstico, los valores de los anticuerpos anticitoplasma de los
neutrófilos (ANCA) eran de 1/60 y los anticuerpos anti-ADN, anti-proteinasa 3,
anti-mieloperoxidasa, anti-SSA/Ro-52, anti-Jo-1, anti-Mi-2, anti-PL-7 y
anti-PL-12, anti-SRP-54, anti-EJ, anti-NXP-2, ANAm y crioglobulinas fueron
negativos. El complemento era normal.
Durante el
último año, tres meses antes del evento en cuestión, sufrió un episodio de
pericarditis y luego se sospechó una miocarditis que finalmente no se confirmó
mediante estudios por imágenes.
Había
recibido varias líneas terapéuticas por el mal control de los síntomas. En los
últimos dos meses, había sido tratada con infliximab y metotrexato.
Acudió al
Servicio de Urgencias por dolor torácico opresivo de esfuerzo de una semana de
evolución que, en las últimas 72 h, ocurría en reposo. Se realizó un
electrocardiograma que mostró un descenso del segmento ST en I, II y de V3 a
V6, con elevación en aVR y cambios dinámicos durante los episodios de angina.
El
ecocardiograma reveló una FEVI preservada con hipocinesia septal e inferior,
más acentuada en segmentos basales, estructura y función del ventrículo derecho
normales, y ausencia de valvulopatías significativas y derrame pericárdico.
Dados los
cambios dinámicos del segmento ST durante los episodios de angina, se realizó
un cateterismo emergente que mostró estenosis ostial severa del tronco
coronario izquierdo con luminograma liso, sugerente de compresión extrínseca
del vaso. No se consiguió cateterizar la arteria coronaria derecha, aunque, por
aortografía, se visualizó tardíamente el segmento medio-distal, aparentemente
de buen calibre y desarrollo, y con circulación heterocoronaria. El resto de
los vasos epicárdicos no tenía lesiones significativas (Figura 1).
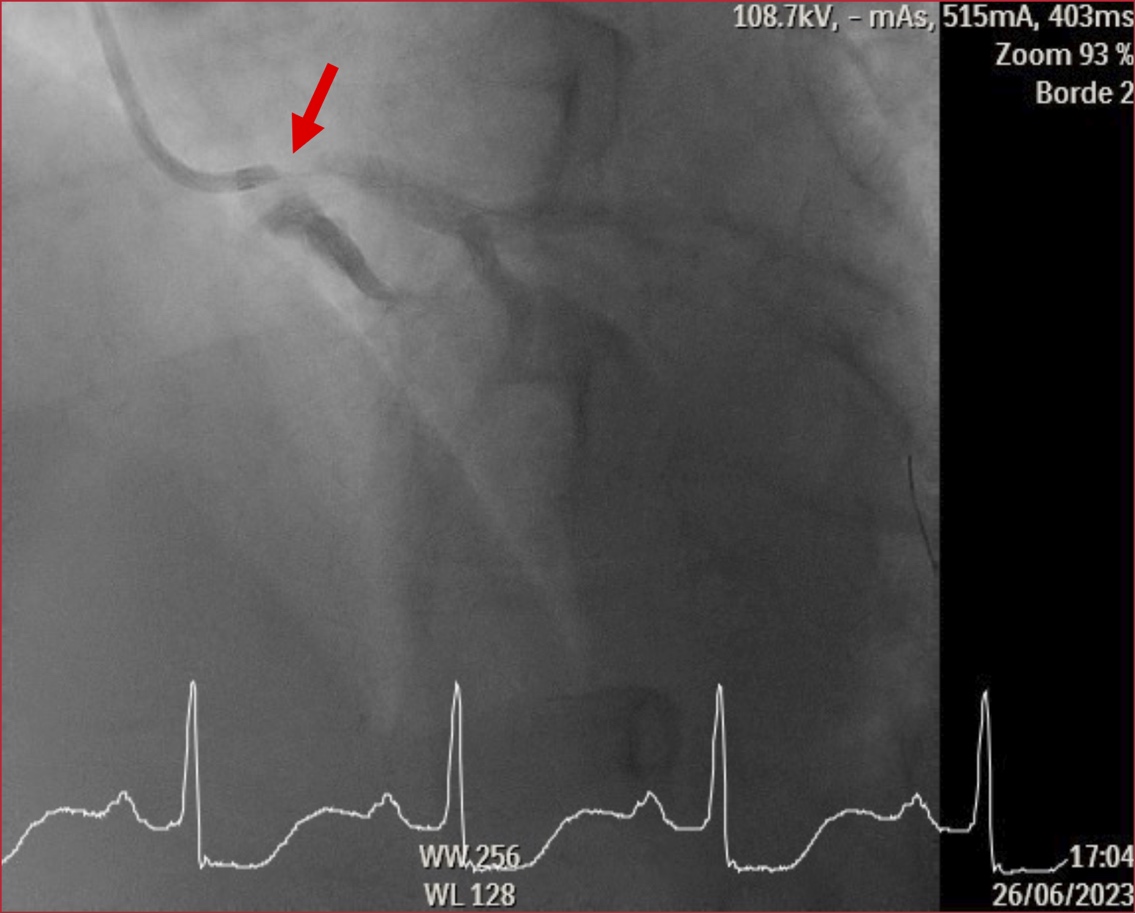
Figura 1. Estenosis ostial severa del tronco coronario izquierdo, sugerente de compresión extrínseca.
Ante los
hallazgos angiográficos y los antecedentes de la paciente, se decidió finalizar
el procedimiento sin llevar a cabo la revascularización y solicitar una
tomografía computarizada para registrar la anatomía coronaria y valorar una
condición aórtica concomitante. La tomografía mostró (Figura 2):
• Signos de aortitis, se visualizó el
engrosamiento concéntrico de la pared vascular aórtica con tenue hiperdensidad
que sugiere compromiso inflamatorio.
• Un defecto de repleción en el origen de
la arteria coronaria derecha con flujo distal, así como estenosis en el origen
del tronco coronario izquierdo con flujo distal.
• Un defecto de repleción en el origen de
la arteria subclavia izquierda y, también, en el origen de la arteria ilíaca
común izquierda, con repermeabilización distal de ambas.
• Estenosis en el origen de la arteria
renal izquierda con buen flujo posterior.
• Un marcado aumento del calibre del
tronco arterial pulmonar, de hasta unos 42 mm, que sugiere hipertensión
pulmonar.
• Un leve engrosamiento pericárdico
ligeramente hiperdenso, en posible relación con pericarditis.
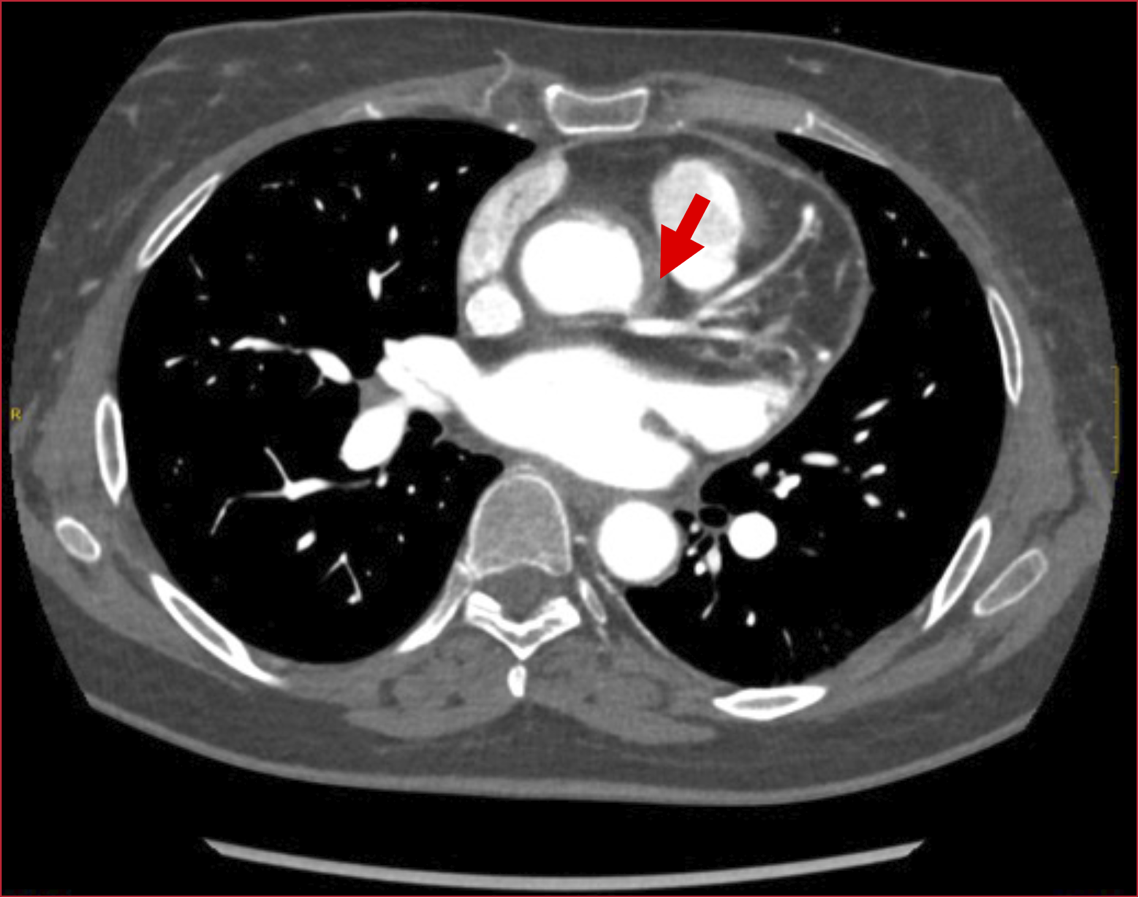
Figura 2. Aortitis. Estenosis del tronco coronario Izquierdo con flujo distal.
Ante dichos
hallazgos, se administró metilprednisolona 500 mg, por vía intravenosa, y se
contactó con el Servicio de Cirugía Cardíaca dada la posible necesidad de
revascularización quirúrgica, por lo que fue trasladada a la Unidad de Cuidados
Intensivos de nuestro hospital.
A su llegada,
fue evaluada por un equipo multidisciplinario compuesto por los médicos de los
Servicios de Medicina Intensiva, Cirugía Cardíaca y Medicina Interna. Se
descartó el tratamiento quirúrgico por la inflamación sistémica y difusa a
nivel vascular. Se decidió iniciar un tratamiento inmunosupresor y
antinflamatorio con metilprednisolona 1 g, por vía intravenosa, durante 5 días;
metotrexato 20 mg, por vía subcutánea, semanal y rituximab 1 g, por vía
intravenosa, cada 15 días. En los períodos de descompensación de la enfermedad,
aumentaron los valores de la velocidad de sedimentación globular a 53 mm,
proteína C reactiva hasta 42,9 mg/l y fracciones del complemento C3 y C4 a 178
y 53,9 mg/dl, respectivamente.
Durante las
primeras 36 h, la paciente sufrió hasta tres episodios de angina con los mismos
cambios dinámicos electrocardiográficos y elevación de troponina ultrasensible
de hasta 5,722 ng/l, acompañados, en alguno de ellos, de signos de
insuficiencia cardíaca con compromiso respiratorio (taquipnea y desaturación)
que obligaron a aumentar los requerimientos de oxigenoterapia y que remitieron
tras el tratamiento vasodilatador con nitroglicerina por vía intravenosa en
perfusión continua a dosis de hasta 4 mg/h y bolos de morfina.
Ante la
persistencia de los síntomas, se procedió a reevaluar, junto con la sección de
Autoinmunes de los Servicios de Medicina Interna, Hemodinámica y Cirugía
Cardíaca, la posibilidad de realizar una angioplastia con colocación de
endoprótesis sobre el tronco coronario izquierdo, que se consideró factible,
pero que finalmente, no se realizó por la posibilidad de que surgieran
complicaciones de difícil solución quirúrgica, y se optó por intensificar el
tratamiento médico inmunodepresor para lo cual se sustituyó el rituximab por
ciclofosfamida (1,5 g mensual) por tener un efecto antinflamatorio más precoz,
se mantuvo el tratamiento con corticoides y se asoció doble antiagregación con
ácido acetilsalicílico y tirofibán, más anticoagulación con heparina sódica en
perfusión continua. Además, se optimizó el tratamiento antianginoso con
betabloqueadores.
A las 24 h de
iniciar el nuevo tratamiento médico, la paciente tuvo una buena respuesta y se
mantuvo asintomática, por lo que se suspendió la anticoagulación y la perfusión
de tirofibán.
Finalmente,
tras siete días en la Unidad de Cuidados Intensivos, es dada de alta al
Servicio de Medicina Interna con nitroglicerina transdérmica, prednisona por
vía oral en pauta descendente, betabloqueadores y antiagregación simple con
ácido acetilsalicílico 100 mg/día.
La paciente
permaneció sin síntomas en planta de hospitalización y fue dada de alta a los
pocos días para continuar su tratamiento y seguimiento de manera ambulatoria.
Ante la recurrencia de episodios de angina, finalmente, se decidió el
tratamiento quirúrgico una vez controlada la inflamación, y se realizó una
triple revascularización coronaria programada, fue dada de alta al domicilio a
la semana, sin complicaciones en el posoperatorio inmediato.
Discusión
El síndrome
de Cogan afecta a ambos sexos por igual, la edad promedio de aparición es a los
38 años (36 en mujeres y 40 en hombres).1 Además, puede manifestarse tanto en niños como en
adultos >60 años.2 Se han sugerido diferentes factores
desencadenantes, como infecciones de la vía respiratoria superior y el
tabaquismo.1-4 Sin
embargo, la causa subyacente aún no está claramente definida.
El síndrome
se manifiesta con síntomas progresivos inflamatorios tanto oculares como
vestibulococleares, con la posibilidad de manifestaciones sistémicas que
afectan principalmente a vasos de gran tamaño. Los síntomas oculares y
vestibulococleares suelen desarrollarse en días o semanas, aunque, en
ocasiones, pueden tardar meses en aparecer, lo que dificulta y retrasa el
diagnóstico.2,3 Los
síntomas auditivos se asemejan a los del síndrome de Ménière e incluyen
vértigo, náuseas, vómitos, tinnitus y pérdida auditiva, y esta es la forma más
común de presentación hasta en el 50% de los pacientes.1,2,4 La principal manifestación ocular es
la queratitis intersticial que provoca enrojecimiento ocular, dolor, fotofobia
y visión borrosa.
El 15-20% de
los pacientes tiene manifestaciones sistémicas que pueden simular una aortitis
o una vasculitis que afecta a vasos grandes o medianos. La aortitis puede
desarrollarse semanas o años después del inicio de la enfermedad, provocando
dilatación de la aorta proximal, insuficiencia aórtica, arteritis coronaria,
infarto de miocardio, pericarditis, hipertrofia del ventrículo izquierdo y
arritmias. En el caso de actuar como una vasculitis de vasos grandes, puede
afectar a las principales ramas de la aorta, así como a vasos mesentéricos o
renales. Las vasculitis de vasos de tamaño mediano pueden causar sangrado
gastrointestinal, proteinuria, microhematuria, vasculitis cutánea y, en casos
más graves, manifestaciones neurológicas. Las manifestaciones sistémicas
inespecíficas, como fiebre, pérdida de peso, fatiga, dolor musculoesquelético y
cefalea, también son comunes.1-4
El
diagnóstico se basa en datos clínicos, como los síntomas antes descritos, y en
los hallazgos de los análisis clínicos, como anemia, elevación de la velocidad
de sedimentación globular y de la proteína C reactiva o leucocitosis.1-3 Es necesario descartar otras
entidades, como infecciones (sífilis, tuberculosis o por clamidia) y otras
causas inflamatorias (poliarteritis nodosa, granulomatosis con poliangeítis y
enfermedad de Takayasu, principalmente). En todos los casos después del diagnóstico,
el ecocardiograma es obligatorio para evaluar si hay aortitis e insuficiencia
aórtica. Si hay dolor concordante con enfermedad coronaria, la coronariografía
estaría indicada.4 Hasta el momento, no existen pruebas
específicas para el diagnóstico.
No se han
llevado a cabo ensayos clínicos aleatorizados que comparen distintos regímenes
de inmunosupresores para tratar el síndrome de Cogan. Por lo general, la
elección del tratamiento se basa en series de casos y enfoques utilizados para
otras enfermedades autoinmunes. La intensidad del tratamiento se ajusta según
la gravedad del cuadro. Los corticoides tópicos pueden ser suficientes para las
manifestaciones oculares moderadas, mientras que el tratamiento sistémico es
necesario si hay pérdida de la audición, afecciones oculares graves o
manifestaciones sistémicas. El tratamiento típico incluye prednisona (0,5-2
mg/kg/día) con disminución gradual durante 2-4 semanas, si hay mejoría clínica.
También se ha comunicado la administración de dosis iniciales altas de
metilprednisolona durante 1-5 días. Si no hay respuesta, no es posible reducir
la dosis de corticoides o surgen complicaciones relacionadas con su uso, se
pueden añadir otros agentes inmunosupresores, como metotrexato, azatioprina,
ciclofosfamida, ciclosporina, infliximab, etambutol, etanercept, rituximab,
tocilizumab o tacrolimus.1-5 Aunque
los resultados de estos tratamientos en ciertos pacientes son prometedores, la
limitada información sobre su eficacia impide la extrapolación de conclusiones
sólidas. En casos de insuficiencia aórtica que no mejora con el tratamiento
médico, puede ser necesario recurrir a la reparación, preferiblemente cuando la
enfermedad está en remisión.4
La mayoría de
los pacientes experimentarán recaídas a lo largo de la enfermedad y, en las
series de casos revisadas, las muertes se produjeron directa o indirectamente
como resultados de las manifestaciones de la enfermedad o de su tratamiento,
como roturas de aneurismas de aorta, amiloidosis, sangrado gastrointestinal o
infartos de miocardio.1-4
Conclusiones
El síndrome
de Cogan es una entidad poco frecuente que, en la actualidad, no tiene un
régimen terapéutico claramente establecido, por lo que este se basa en los que
se administran para otras enfermedades autoinmunes. Los pacientes con
manifestaciones sistémicas con implicación de grandes vasos tienen una elevada
tasa de mortalidad y no está clara la indicación ni el momento óptimo para el
manejo quirúrgico/invasivo. En la paciente presentada, se llevó a cabo una
revascularización miocárdica quirúrgica con buen resultado tras la persistencia
de los síntomas, pese al tratamiento inmunosupresor. Se necesitan más estudios
para establecer el tratamiento óptimo y las indicaciones de cirugía en estos
pacientes.
Bibliografía
1.
Gluth MB,
Baratz KH, Matteson EL, Driscoll CLW. Cogan syndrome: A retrospective review of
60 patients throughout a half century. Mayo Clin Proc 2006; 81(4):
483-488. https://doi.org/10.4065/81.4.483
2.
Mazlumzadeh
M, Matteson EL. Cogan’s syndrome: An audiovestibular, ocular, and systemic
autoimmune disease. Rheum Dis Clin North Am 2007; 33(4): 855-874. https://doi.org/10.1016/j.rdc.2007.07.015
3.
Tayer-Shifman
OE, Ilan O, Tovi H, Tal Y. Cogan’s
syndrome – clinical guidelines and novel therapeutic approaches.
Clin Rev Allergy Immunol 2014; 47(1): 65-72. https://doi.org/10.1007/s12016-013-8406-7
4.
Grasland
A. Typical and atypical Cogan’s
syndrome: 32 cases and review of the literature. Rheumatology 2004; 43(8):
1007-1015. https://doi.org/10.1093/rheumatology/keh228
5.
Unizony
S, Stone JH, Stone JR. New treatment strategies in large-vessel vasculitis. Curr Opin
Rheumatol 2013; 25(1): 3-9. https://doi.org/10.1097/bor.0b013e32835b133a